
Andrew Jesaitis




In 2011 Golden Helix and Expression Analysis joined forces to build a cloud based RNA Seq pipeline. Upon joining Golden Helix as a Full Stack Developer I was able to work on this platform. Ultimately, it was opened as a Beta in mid-2012 and publicly released at the end of 2012.
The idea behind RNA-Seq analysis is to determine the expression level of a gene in a subject’s body. This level can be inferred through the sequencing of mRNA, the product of transcribed DNA. A single gene may be transcribed many times to create more mRNA and ultimately more protein. Thus, if we can measure the relative levels of mRNA we have some basis for estimating how much protein will be present. This is crucial information since many genetic disorders are not all or nothing. Sometimes a person can do fine if one copy of a gene doesn’t produce protein (they are said to to haplosufficient). On the other hand the creation of too much protein can problematic.
To measure the levels of RNA, you would collect a sample and sequence the RNA. After the raw sequence data has been created, you’d need a tool to make sense of the data. That is where the RNA-Seq pipeline would be employed.
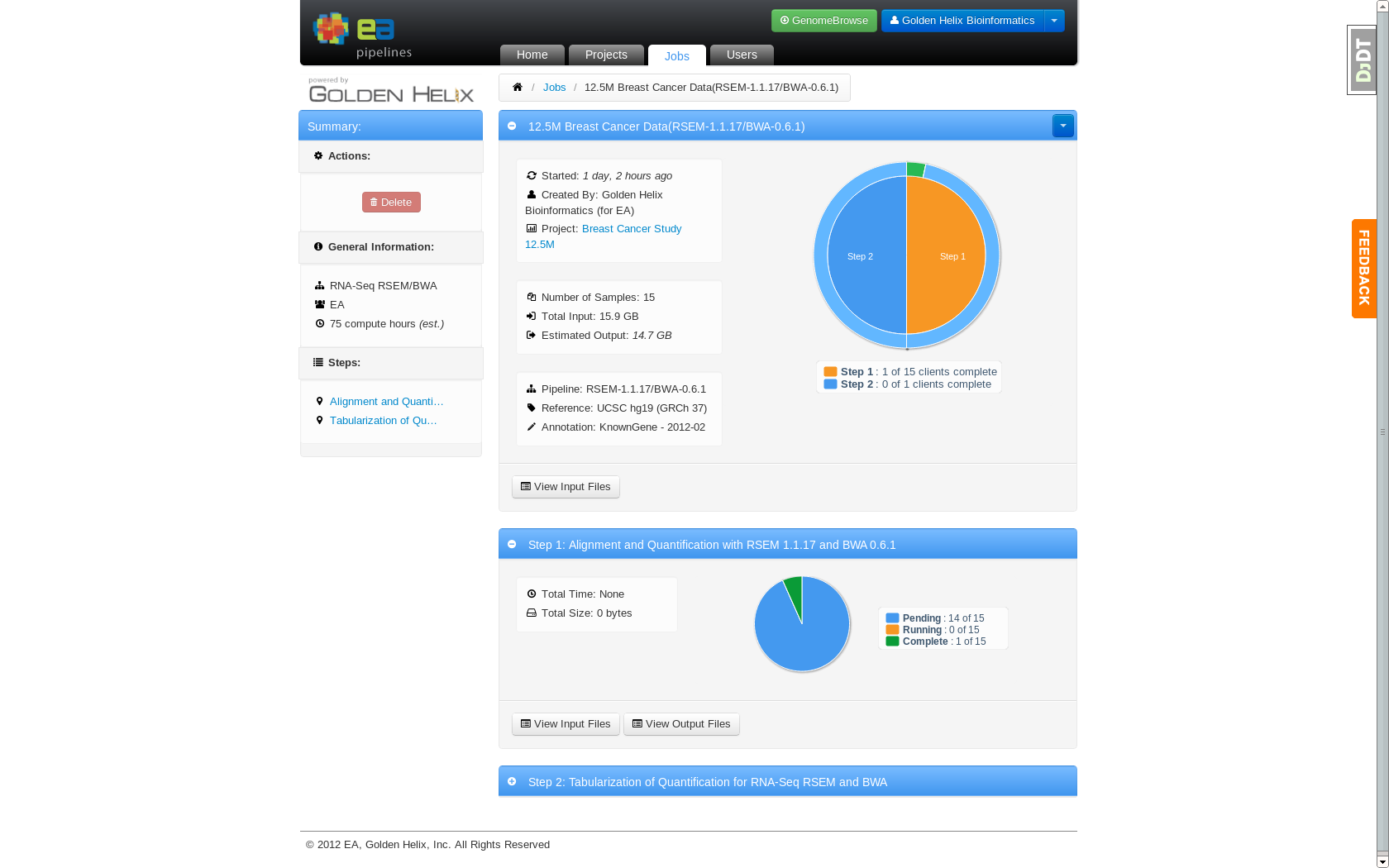
Job detail page showing an in progress job running on 15 nodes
The sequence data comes in as short random reads. These look like 30-200 character strings and are contained in a “Fastq” file. These reads are first aligned to the human reference sequence. A sample may have millions of these reads and they must be matched to the billions of bases in the human genome. Obviously, this is a compute intensive process and is a perfect match for cloud-based machines.
After the reads have been aligned, the read depth must be “quantified”. Certain areas of the genome will have many reads aligning to the region. These are transcripts, which are portions of genome that code for protein. Since we already know the sequences of these regions, we measure the coverage to determine the expression level. Next, we estimate the relative concentration of these reads between samples. Although the quantification is not as compute intensive as alignment, it is still nice to do it cloud-side as it is a repetitive bioinformatic task and can still require a fair amount of compute resources.
Finally, we tabularize these results into a format that could easily be analyze in Golden Helix’s Snp and Variation Suite or in one of the many R packages used in Differential Expression Analysis.
The bioinformatics pipeline is user customizable. The end user can select to use BWA or CASSAVA for alignment and either Tophat-Cufflinks or RSEM for quantification. Additionally, the user can select what assembly to use and what source should be used for transcript annotation. These parameters can be specified on the job creation page. The specific pipeline command line options were hidden from the user and defined by the bioinformatics team at EA.
Surrounding the ability to run user selectable pipelines is a large amount infrastructure.
The stack consists of a variety of tools including:
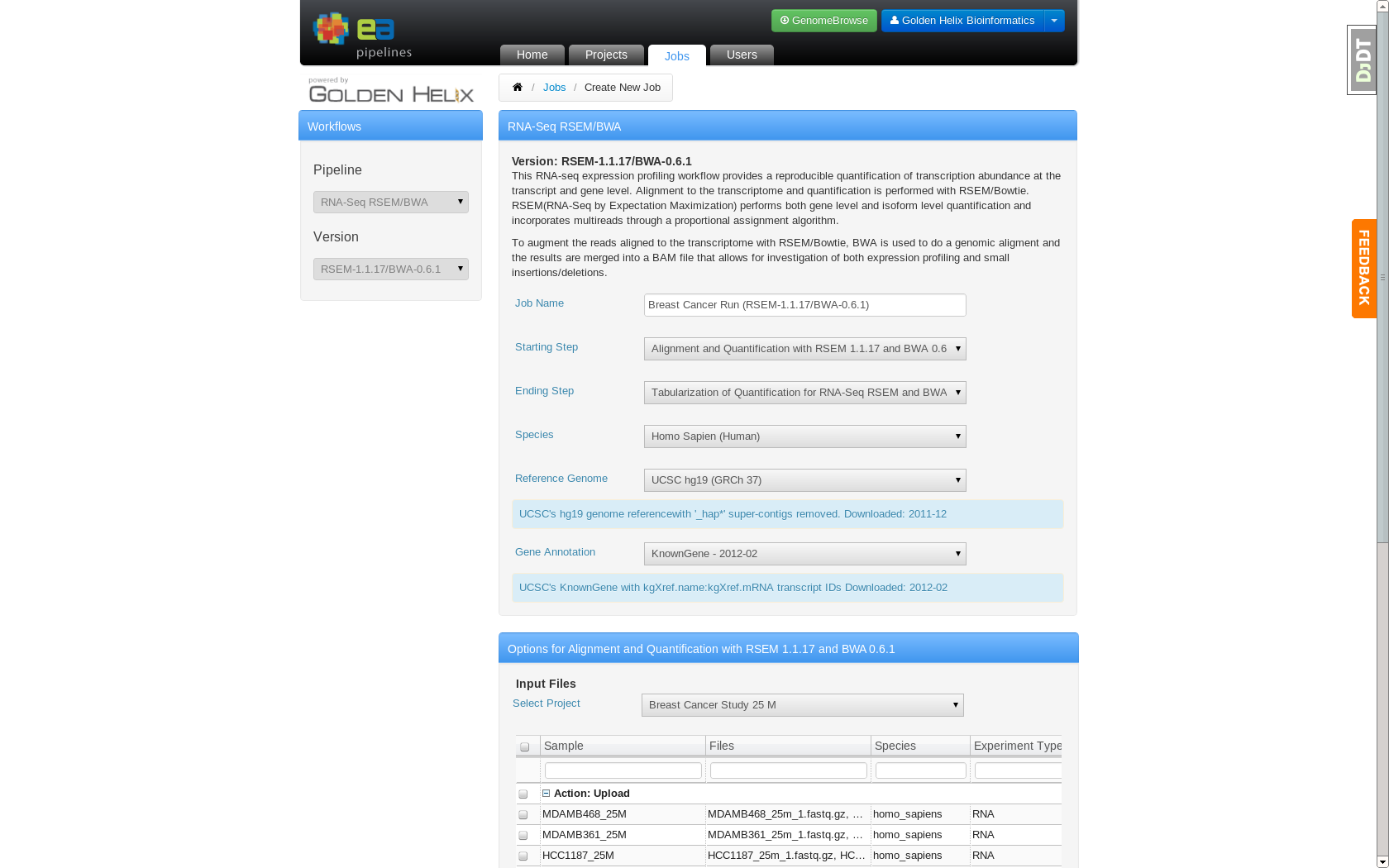
Job Creation page which allows users to specify analysis settings
Server-Side:
Client Side:
It resides on on AWS and uses Boto coordinate services:
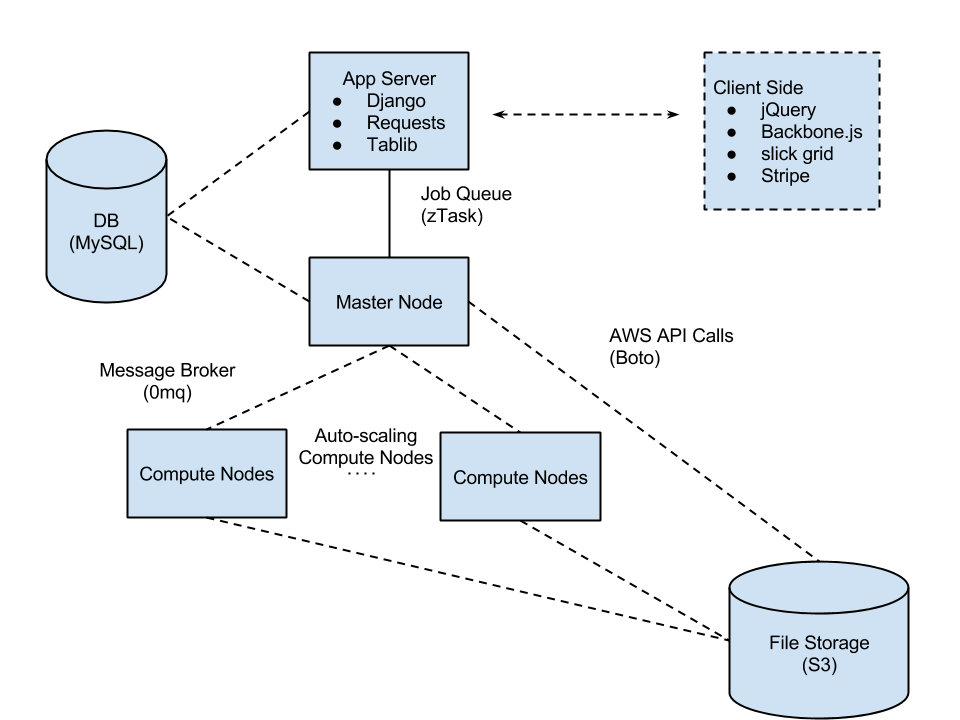
Simplified view of the basic architecture of the system
And we employ the following deployment and maintenance tools:
The architecture is flexible enough to be adapted to run DNA alignment and variant calling, genotype phasing, and other bioinformatic tasks.
My role included the implementation of the user management system, billing and charging system and job creation UI. I also built a desktop multi part unloader to allow users to move hundreds of gigabytes of data to S3 easily. This tool was written in python using PyQt bindings for the UI.
Later I’ve worked on all parts of the system ranging from Bash scripting for bionifomatics on the compute nodes, data curation, the job queue and messaging system, file management system and administrative management system for running and inspecting compute nodes.